
+91 6002993949
submission@iarconsortium.org
Open Access
ISSN (Print) : 2708-6259
ISSN (Online) : 2708-6267
Genome assembly is a critical process in bioinformatics, where short DNA sequences (reads) are pieced together to reconstruct an organism's genome. As sequencing technologies generate vast amounts of data, the need for effective computational methods to assemble these reads has grown. Two prominent graph theory-based approaches—the overlap graph and the Hamiltonian path approaches—offer different strategies for this task. This study focuses on the construction and decoding aspects of these graph-based methods, providing a comparative analysis of their effectiveness in genome assembly. This research will explore the intricacies of constructing and decoding these graphs, examining how each approach handles challenges such as repetitive sequences, sequencing errors, and varying read lengths. The construction phase will be analysed in terms of computational efficiency, focusing on the algorithms used to build the graphs and the preprocessing required to manage large datasets. The decoding phase will be evaluated based on the accuracy of the assembled genome, considering factors like contiguity (N50), error rates, and the ability to resolve complex genomic regions. A key aspect of this research is the comparison of the decoding strategies used in both approaches. For the overlap graph approach, the focus will be on greedy algorithms that iteratively connect reads with the best overlaps. The Hamiltonian path approach, on the other hand, will be examined through the lens of heuristic and approximation algorithms designed to tackle its inherent computational complexity. This research will be supported by practical experiments using real-world sequencing data, allowing for a detailed evaluation of how each method performs under different conditions. The research will also consider the scalability of these approaches, particularly in the context of emerging sequencing technologies that produce longer and more accurate reads. Ultimately, this thesis aims to provide a clear understanding of the trade-offs involved in the construction and decoding of overlap graphs and Hamiltonian paths for genome assembly. By focusing on the graph theory aspects of these methods, the research will offer insights into their strengths and limitations, guiding the selection of the most appropriate approach for different genomic challenges. The findings will contribute to the ongoing development of more efficient and accurate genome assembly techniques, with potential applications in a wide range of biological and medical research.
Genome assembly is a critical process in bioinformatics, aimed at reconstructing the complete genome from short DNA sequences (reads) produced by sequencing technologies. As sequencing technologies have advanced, the volume of data has significantly increased, necessitating effective computational methods for genome assembly. Two prominent graph- based approaches in this field are the overlap graph approach and the Hamiltonian path approach. This literature review focuses on the construction and decoding aspects of these methods, providing a comparative analysis of their effectiveness. The overlap graph approach has been widely utilized in genome assembly, particularly with the advent of next-generation sequencing technologies. In this method, each read is represented as a vertex in a graph, with edges connecting vertices that exhibit significant overlaps. The primary goal is to decode the graph by finding a path that maximizes these overlaps to reconstruct the genome sequence. This approach gained prominence with the development of the de Bruijn graph [1], which simplified the assembly process by breaking down reads into k-mers and connecting them based on overlaps. This method proved effective for bacterial and viral genomes but has faced challenges with more complex genomes due to repetitive sequences and ambiguous overlaps.
Enhancements to the overlap graph approach have focused on improving computational efficiency and handling repetitive sequences. [2] introduced the string graph, which simplifies the overlap graph by removing redundant edges, thereby reducing complexity and improving scalability. Despite these advancements, the overlap graph approach still struggles with repetitive sequences and computational efficiency, particularly when dealing with large and complex genomes. Recent advances in sequencing technologies, such as long- read sequencing, have mitigated some of these challenges but have not completely resolved the limitations of the overlap graph approach.
In contrast, the Hamiltonian path approach represents a more theoretically powerful method but is less commonly used due to its computational complexity. This approach constructs a graph where each read is a vertex, and the goal is to find a Hamiltonian path that visits each vertex exactly once. While this can theoretically provide a highly accurate assembly by ensuring that each read is uniquely placed, the Hamiltonian path problem is NP-hard, making it computationally intensive for large datasets [3]. Early work in applying this approach to genome assembly was limited by high computational costs. However, theoretical studies have shown that if a Hamiltonian path can be found, it results in a highly accurate assembly. To address the computational challenges of the Hamiltonian path approach, several heuristic and approximation algorithms have been developed. [4] proposed a greedy algorithm for constructing a Hamiltonian path by iteratively adding edges that extend the current path. More recent research has focused on hybrid approaches that combine elements of both the overlap graph and Hamiltonian path methods to improve accuracy and efficiency. With the advent of long-read sequencing technologies, there has been renewed interest in the Hamiltonian path approach, as longer reads can reduce the complexity of finding a Hamiltonian path by providing more unique overlaps [5] .
The comparative analysis of these approaches highlights their respective strengths and limitations. The overlap graph approach tends to be more computationally efficient and practical for a wide range of genomes, particularly with the use of optimized algorithms like the string graph. However, it can result in fragmented assemblies in genomes with high repeat content. Conversely, the Hamiltonian path approach offers potential advantages in terms of assembly contiguity but is constrained by its computational complexity and the difficulty of solving the NP-hard problem for large datasets [6] . Hybrid strategies that leverage the strengths of both approaches may provide a more robust solution; particularly as sequencing technologies continue to advance [7]. Overlap graphs have been used extensively in genome sequencing, with applications in both prokaryotic and eukaryotic genomes. In prokaryotic genomes, overlap graph assembly is often used to generate high-quality, closed genomes from short-read sequencing data [8]. In eukaryotic genomes, long-read sequencing technologies such as PacBio and
Oxford Nanopore have enabled the construction of more complex overlap graphs that can span repetitive regions and structural variations [9].
An Overlap Graph (OG), in the context of a set of finite words, can be described as a complete weighted directed graph where each word is represented as a node, and the weight of each arc corresponds to the length of the longest overlap from one word to another. This overlap is inherently asymmetric. In essence, an OG is a complete directed graph, weighted on its arcs, with nodes representing the words in the set, and the weight of an arc (u, v) equalling the length of the maximum overlap from string u to string v. The Overlap Graph is instrumental in reconstructing genome fragments or computing the shortest superstrings, which serve as a compressed representation of the input data.
However, the Overlap Graph requires quadratic space relative to the number of words, which imposes limitations on its scalability. To address this, the Hierarchical Overlap Graph (HOG) has been proposed as an alternative. The HOG encodes all maximal overlaps but operates with space complexity that is linear in the sum of the lengths of the input words. Constructing this graph involves calculating the weights of the arcs by solving the All-Pairs Suffix Prefix (APSP) overlaps problem on the set of words. Overlap graphs can be generated by aligning reads to a reference genome or through de novo assembly methods that create a graph from the ground up. Once the overlap graph is constructed, it can be traversed to generate a consensus sequence representing the complete genome.
One of the significant challenges in overlap graph assembly is managing sequencing errors, which can introduce false overlaps and complicate the graph structure. To mitigate this, various methods have been developed to filter out low-quality reads and correct sequencing errors prior to constructing the overlap graph, ensuring a more accurate and reliable assembly.
In Overlap graph each node is a read e.g., GCTCTAGCCCCTCATTT. Therefore, we draw a directed edge A → B only when the suffix on A overlaps prefix of B.
Figure 1: Example of prefix and suffix overlap
Example 1: Let G ={TACGAT, GTACGT, ACGTAC, GTACGA, CGTACG, TACGTA} be a
multiset of all 6-mers from long nucleotides of a genome sequence and the edges be overlaps of length 4. For the threshold of this example, the suffix/prefix match will be an exact match and has a length of at least 4.
Figure 2: First Overlap
Figure 3: Second Overlap
Figure 4: Third Overlap
Figure 5: Forth Overlap
The diagrams above (see Figure 2 to Figure 5) illustrate how prefix to suffix overlaps occurs through each node as long as overlaps of length ≥4. Each node is a distinct nucleotide sequence from a genome read and by using an edge connecting all distinct node that have overlaps will result in the below diagrams. From the diagrams above the weight on each node is shown above, some node has weight of 4 and most have of weight 5.
Figure 6: Completed OG of G
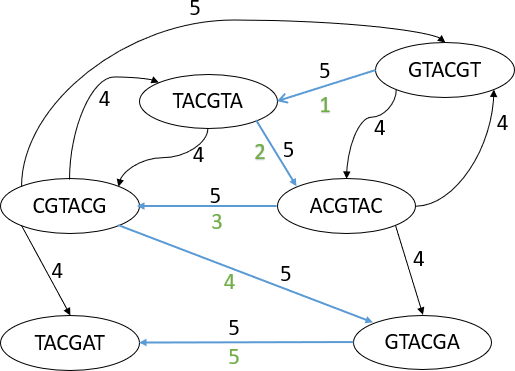
Figure 7: Completed OG of G with Path line
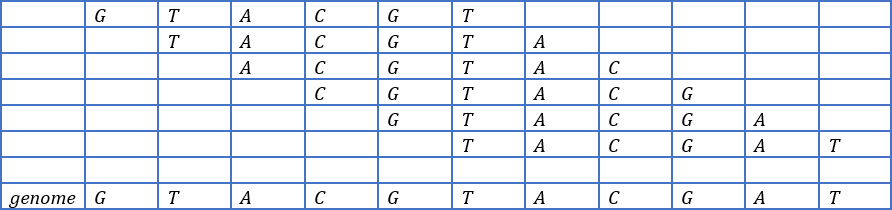
After completing connecting the 6-mers reads for this Overlap graph, at the end we’ll come out with the diagram above (see Figure 6) clearly showing all the overlaps nodes joined together with a directed edge from prefix to suffix. The weight of this Overlap graph is shown on either the right side or the top part of the edge. Finally, for reconstructing the original genome, we walk through the overlap graph following each directed edge creating a path. This walk is shown above(see Figure 7), where we can deduce the original genome sequence for our genome. The walk will result in the table below(see Table 1). In conclusion, overlap graphs are a powerful tool in genome sequencing, allowing the assembly of complete genomes from overlapping reads of DNA fragments. Their application extends beyond genome sequencing to other genomic applications such as metagenomics and transcriptomics, making them a versatile and valuable tool in genomic research.
From the table (see Table 1) we can clearly see from the walk and reconstruct the original genome sequence from the overlaps as GTACGTACGAT. The OG has several drawbacks. First, it is not possible to know whether two distinct arcs represent the same overlap. Second, the OG has an inherently quadratic size since it contains an arc for each possible (directed) pairs of words. Overlap graphs have been a popular approach to genome assembly for many years, but they have limitations when it comes to handling repeat regions and errors in the data. To address these limitations, researchers have recently turned to Hamiltonian graph-based approaches. Subsequent research has explored different variations of the string graph algorithm and other Hamiltonian path-based approaches for genome assembly. For example, the "de Bruijn graph" algorithm, which was previously mentioned as an alternative to overlap graphs, can also be viewed as a type of Hamiltonian graph.
In this section, we show how the genome is sequenced using Hamiltonian approach. If there is a closed walk in a connected graph that passes every vertex of the graph exactly once, with the exception of the root or starting vertex, the graph is referred to as a Hamiltonian graph. The Hamiltonian walk must not repeat any edges. Another definition of a Hamiltonian graph states that if a graph is connected and Hamiltonian circuit exists, the graph in question is said to be a Hamiltonian graph. A set of points called the vertex is what makes up a graph. These points are connected by a set of lines called the edges. An exact one-time pass through each vertex characterizes a Hamiltonian walk-in graph G.
These theorems provide conditions that guarantee the existence of a Hamiltonian cycle in a graph, which can be used to solve practical problems related to graph theory.
Objective: Use overlapping DNA reads in order to reconstruct the original genome sequence.
When having our fragments of the genome they often overlap. We are able to make use of this overlap and stitch them together. Assuming our fragments (often referred as mers) are 3 molecules long (3-mer). For instance, we could have fragments such as AAT, GCG, CAA. By also assuming they overlap with two molecules. This means the fragment AAT must be followed by a fragment beginning with AT e.g., ATT.
We create a Hamiltonian graph where each node is a fragment. And there is an edge going from a node to another when they only overlap by two nucleotides bases. So, the node AAT would have an edge connecting it to ATT.
Example 2: Let S= {AAT, GCG, GCA, ATG, TGG, TGC, GGC, GTG, CGT, CAA} be a
multiset of all 3-long nucleotides of a genome sequence. By constructing a network that represents the overlap information in our reads. Each k-mer nucleotide from the multiset becomes a vertex( see Figure 8) ; two vertices will be connected by a directed vertex if and only if the k-1 rightmost nucleotides from the first vertex overlap with the k-1 leftmost nucleotides of the second vertex.
First, we create a node for each read e.g. GTG. Prefix: First two nucleotide of a read (GTG). Suffix: Last two nucleotide of a read (GTG). Note: Different 3-mers can share a prefix/suffix: CTG ,ATG, TGA.
Figure 8: Aligning the 3-mer nodes.
As shown above (see Figure 8), DNA reads are aligned and ready to be joined using overlap reads from prefix to suffix.
Figure 9: Connecting DNA nodes based on prefix and suffix
The Figure 9 shows how to connect these DNA nodes based on the prefix and suffix. As illustrated from diagram above (see Figure 9), with nodes ATG and GTG connecting to nodes TGC and TGG based on the overlapping part of the nucleotide. In the Hamiltonian approach, the problem of genome sequencing is transformed into a mathematical problem of finding the minimum cost path in a graph. The graph represents the overlap of the reads obtained from the sequencing process. The cost of the path is usually defined as the sum of the weights of the edges of the path. The weights of the edges correspond to the number of nucleotides matches between the two reads connected by the edge.
From Figure 10 , we can clearly see a completed graph when connecting all nodes with the same prefix to the node with the same suffix and shows how the genome sequencing is done using the Hamiltonian approach. Now we need to deduce the order of the DNA and for us to do so we need to follow the path on the diagram that takes us from where we started.
Our Hamiltonian cycle will be:
ATG→TGG→GGC→GCG→CGT→GTG→TGC→GCA→CAA→AAT→ATG.
Therefore, our genome based on this reconstruction is ATGGCGTGCAAT
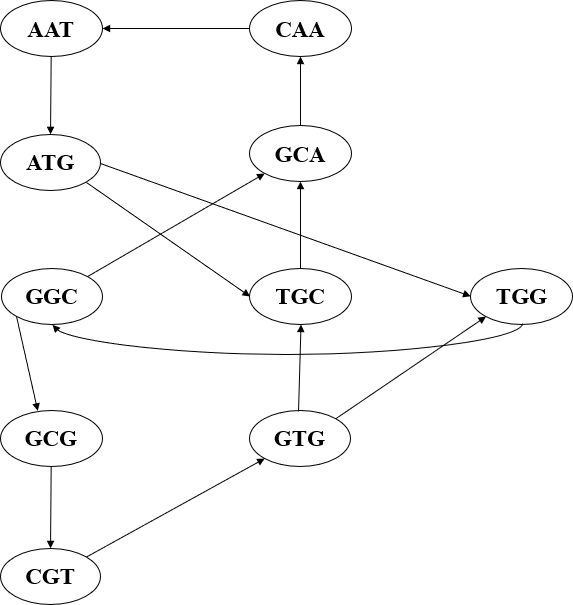
Figure 10: complete Hamiltonian Graph of example
One of the earliest works on the Hamiltonian approach in genome sequencing was proposed by Pevzner et al. They proposed an algorithm called "Haplotyping as Maximum Parsimony" (HAMPATH) for haplotype assembly. The algorithm used a Hamiltonian path approach to solve the haplotype assembly problem. The algorithm was shown to be effective on simulated and real data.
In a later work, Pevzner et al. proposed a new algorithm called "EULER" for genome assembly using the Hamiltonian approach. The algorithm was based on finding Eulerian paths in the overlap graph of the reads. The authors showed that the algorithm was able to handle sequencing errors and was more accurate than other assembly methods. Li et al. proposed an algorithm called "HGAP" for hierarchical genome assembly using the Hamiltonian approach. The algorithm used a hierarchical approach to assemble long reads into contigs and scaffolds. The authors showed that the algorithm was able to assemble the genome of E. coli with a single contig, which was the first time this had been achieved.Recently, Ren et al. proposed a new algorithm called "HINGE" for haplotype assembly using the Hamiltonian approach. The algorithm was shown to be highly accurate and efficient on both simulated and real data. The authors also compared their algorithm with other state-of- the-art algorithms and showed that their algorithm outperformed them in terms of accuracy and efficiency. Another example is the use of the Hamiltonian approach to analyze the folding of DNA. By treating DNA as a polymer chain, researchers have been able to use Hamiltonian models to study the thermodynamics of DNA folding and the effect of various factors, such as histone modifications and DNA-binding proteins, on this process.
The comparative analysis of the Overlap Graph (OG) and Hamiltonian Path (HP) approaches in genome assembly reveals both similarities and differences that are critical to understanding their application and effectiveness in reconstructing genomic sequences.
Both the OG and HP approaches are rooted in graph theory and are fundamental to the field of genome assembly. They each involve representing sequences as nodes within a graph and determining the optimal way to traverse these nodes to reconstruct the genome. Additionally, both methods aim to generate a consensus sequence that accurately represents the genome from a set of short DNA reads, handling the complex problem of assembling sequences from potentially noisy and incomplete data.
Despite these similarities, the approaches differ significantly in their conceptual underpinnings, computational complexity, and practical applications. The OG approach constructs a complete weighted digraph where nodes represent individual reads, and edges correspond to the overlaps between these reads. This method excels in its ability to handle large datasets and is particularly effective when the overlaps are significant, although it can be computationally demanding due to its quadratic space requirements.
On the other hand, the HP approach is more theoretically oriented, focusing on finding a Hamiltonian path that visits each node exactly once, thereby ensuring that each read is uniquely placed in the genome assembly. While this approach can potentially yield highly accurate assemblies, it is computationally intensive due to the NP-hard nature of the Hamiltonian path problem, making it less practical for large genomes without employing approximation algorithms.
In summary, while both the Overlap Graph and Hamiltonian Path approaches are integral to genome assembly, they are suited to different types of challenges within the field. The OG approach is generally more practical for large-scale assemblies, offering efficiency and scalability, particularly when enhanced by methods like string graphs. Conversely, the HP approach, though more computationally demanding, offers the potential for higher accuracy in specific contexts where precise sequence placement is critical. Understanding the strengths and limitations of each approach is essential for selecting the appropriate method for a given genome assembly task, and ongoing advancements in computational algorithms and sequencing technologies may continue to blur the lines between these two methodologies
The authors declare that they have no conflict of interest
No funding sources
The study was approved by the Zhejiang University of Science and Technology, Hangzhou, China
Chaisson, M. J., and G. Tesler. "Mapping Single Molecule Sequencing Reads Using Basic Local Alignment with Successive Refinement (BLASR): Application and Theory." BMC Bioinformatics, vol. 13, no. 1, 2012, p. 238. doi:10.1186/1471-2105-13-238.
Myers, E. W. "The Fragment Assembly String Graph." Bioinformatics, vol. 21, sup. 2, 2005, pp. ii79-ii85. doi:10.1093/bioinformatics/bti202.
Garey, M. R., and D. S. Johnson. Computers and Intractability: A Guide to the Theory of NP-Completeness. W.H. Freeman, 1979.
Pevzner, P. A. "DNA Physical Mapping and Alternating Eulerian Cycles in Colored Graphs." Algorithmica, vol. 13, no. 1-2, 1989, pp. 77-105. doi:10.1007/BF02523481.
Chaisson, M. J., and G. Tesler. "Mapping Single Molecule Sequencing Reads Using Basic Local Alignment with Successive Refinement (BLASR): Application and Theory." BMC Bioinformatics, vol. 13, no. 1, 2012, p. 238. doi:10.1186/1471-2105-13-238.
Koren, S., et al. "Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-mer Weighting and Repeat Separation." Genome Research, vol. 27, no. 5, 2017, pp. 722-736. doi:10.1101/gr.215091.116.
Nagarajan, N., and M. Pop. "Sequence Assembly Demystified." Nature Reviews Genetics, vol. 14, no. 3, 2013, pp. 157-167. doi:10.1038/nrg3398.
Jain, M., H. E. Olsen, B. Paten, and M. Akeson. "The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community." Genome Biology, vol. 17, no. 1, 2016, p. 239. doi:10.1186/s13059-016-1044-5.
Rhoads, A., and K. F. Au. "PacBio Sequencing and Its Applications." Genomics, Proteomics & Bioinformatics, vol. 13, no. 5, 2015, pp. 278-289. doi:10.1016/j.gpb.2015.08.002.
