
+91 6002993949
submission@iarconsortium.org
Open Access
ISSN (Print) : 2789-5998
ISSN (Online) : 2789-6005
In this work, isobutyric acid was converted to ethyl isobutyrate by reacting isobutyric acid in 5 ml H2SO4 and absolute ethanol. Ester has been converted to hydrazide by reacting with hydrazine in absolute ethanol, and finally, the hydrazones formed from the hydrazide interaction with substituted aldehyde were prepared. The aim of evaluating their ability to interact with the 8C7Y protein, which represents the crystal structure of the TYRP1 protein (Tyrosinase-related). Its disorders are associated with skin pigmentation and melanoma. The prepared compounds were diagnosed by physical methods, spectroscopic methods, using infrared spectrum and nuclear magnetic resonance of the proton, as well as the completion of the reaction, and confirmed purity the compounds by thin-layer chromatography. The aim of preparing these compounds is to study molecular docking.
Hydrazones are important chemical type in organic chemistry. Hydrazone derivatives are represented via binding of hydrazine derivatives with aldehyde, ketone substituents. These compounds have received wide attention in recent decades due to their ease of preparation as well as their unique physical and chemical properties, in addition to their diverse application in pharmaceutical fields where hydrazone antimicrobial activity [1], antibacterial [2-4], anticancer [5,6], antioxidation [7], and inflammatory [8], they are also intermediates for the preparation of heterocyclic compounds (oxadiazols, thiadiazols, and triazoles) [9]. Therefore, in this paper, six new hydrazones were prepared with different substituents, and molecular docking [10-12] was studied. The aim of evaluating their ability to interact with 8C7Y protein, which represents the crystal structure of the TYRP1 protein. Its disorders are associated with skin pigmentation and melanoma.
Experimental
Material: The chemicals and solvents were purchased from Fluka, BDH. We verified the spectra IR using a Bruker FT-IR 8400, Proton nuclear magnetic resonance was performed on a Bruker instrument operating at 400 MHz, employing CDCl3 as a solvent and TMS as a standard of chemical shift referencing.
Preparation of Ethyl Isobutyrate (1a)
Mix (0.025) mole of isobutyric acid with 30 mL of absolute ethanol, add with cooling 5 mL concentrated sulfuric acid after that the mixture reflux temperature until completion of the reaction for six hours, monitoring by chromatography of thin layer and the mixture of reaction is equivalent for a solution of sodium bicarbonate diluted by 20%, ether is used to extract the product, the product is dried using calcium chloride, evaporated to give a liquid substance [13,14].
Pale Brown: yield 79%: b.p 86 Co: FT-IR, cm -1: 2939, 2879(CH, CH3), 1734(C=Oester)
H-NMR (CDCl3, 400 MHz) : δ 4.12 (q, 2H), 2.6 (m, 1H), 1.31 (t, 3H), 1.22 (d, 1H).
Preparation of Isobutyric Acid Hydrazide (2a)
Mix (0.01) mole of ethyl iso butyrate with (0.05) mole of hydrazine in (30) ml from absolute ethanol and reflux for six hours. The purity of the compound was confirmed, and following the interaction using the thin layer chromatography technique using a solvent mixture (ethanol, petroleum ether) with a ratio of 3:7. Then evaporated the solvent under rarefied pressure, and the product has been extracted with methylene chloride to give a liquid substance[15] [16].
Brown: yield 88%: b.p 107 Co: FT-IR, cm-1: 3380(N-H), 2944,2832 (CH, CH3), 1680(C=O amide)
H-NMR (CDCl3, 400 MHz) : δ 8.4 (s, 1H, N-H), 2.58(m, 1H), 1.20 (d, 1H)
General procedure for the syntheses of hydrazone (3a-8a)
Mix (0.01) mol of hydrazide prepared with (30) ml ethanol, then add an alcoholic solution of benzaldehyde compound (0.01mol / 25 ml ethanol), the mixture is refluxed in two hours. The reaction has been confirmed to be completed using thin layer chromatography technique using a solvent mixture (ethanol, petroleum ether) with a ratio of 3:7. Then cooled and filtered to give a solid product[17] [18].
N'-(4-hydroxybenzylidene)isobutyrohydrazide: (3a)
Yellow: yield 73%: m.p 278-279 Co: FT-IR, cm-1: 3380(N-H), 2925,2832 (CH, CH3), 3020(ArC-H), 1665(C=O), 1630(C=N)
H1-NMR (CDCl3, 400 MHz) : δ 11.1(s, 1H, N-H ), 9.3(s,1H,O-H), 8.30(s,1H, H-C=N), 7.6-6.8(m, 4H,Ar C-H), 2.6(m, 1H), 1.1(d, 1H)
4-((2-isobutyrylhydrazineylidene)methyl)benzoic acid: (4a)
Yellow: yield 69%: m.p 299 Co: FTIR, cm-1: 3350(N-H ), 2934,2910 (CH, CH3), 3060(ArC-H), 2420-3000(OH), 1681(C=O),1622(C=N)
H1-NMR(CDCl3, 400 MHz) : δ12.1(s,1H,OH), 11.07(s, 1H, N-H ), 8.81(s,1H, H-C=N), 7.9-8.35(m, 4H,Ar C-H), 2.61(m, 1H), 1.12(d, 1H)
N'-(4-nitrobenzylidene)isobutyrohydrazide: (5a)
PaleYellow: yield 70.0%: m.p 307 Co: FT-IR, cm-1: 3445(N-H ), 2923(CH, CH3), 3030(ArC-H), 1699(C=O),1649(C=N), 1344,1551(NO2sym,asym)
H1-NMR(CDCl3, 400 MHz) : δ11.2(s,1H, N-H), 8.58(s,1H, H-C=N), 8.13-8.33(m, 4H,Ar C-H), 2.60(m, 1H), 1.10(d, 1H)
N'-(4-butylbenzylidene)isobutyrohydrazide: (6a)
Orang: yield 75%: m.p 163 Co: FTIR, cm-1: 3420(N-H), 2961,2875(CH, CH3), 3010(ArC-H), 1680(C=O),1647(C=N)
H1-NMR (CDCl3, 400 MHz) : δ 9.5(s, 1H, N-H), 8.41(s, 1H, H-C=N), 7.82-7.21(m, 4H,Ar C-H), 2.80(h, 1H), 1.10(d, 1H), 2.65(t, CH2, 2H ), 1.4(m, CH2, 2H ), 1.13(Hexa, CH2, 2H), 0.92(t, CH3, 3H)
N'-benzylideneisobutyrohydrazide: (7a)
Yellow: yield 71%: m.p 92-94 Co: FT-IR, cm-1: 3356(N-H), 2974(CH, CH3), 3090(ArC-H), 1647(C=O),1624(C=N)
H1-NMR (CDCl3, 400 MHz) : δ 8.31 (s,1H, H-C=N ), 7.72-7.58(m, 4H,Ar C-H ), 2.61(m, 1H), 1.10(d, 1H)
N'-(4-(dimethylamino) benzylidene) isobutyrohydrazide : (8a)
Yellow: yield 85%: m.p 266-267 Co: FT-IR, cm-1: 3444(N-H), 2910(CH, CH3), 3090(ArC-H), 1640(C=O),1623(C=N)
H1-NMR (CDCl3, 400 MHz) : δ 8.45 (s,1H, H-C=N ), 7.31-6.72(m, 4H, Ar C-H ), 3.21(s, 6H, 2CH3), 2.63(m, 1H), 1.12(d, 1H)
Nucleophilic reagent Acid Hydrazide reacted with substituted aldehyde to form Hydrazone (3a-8a), in scheme 1. The prepared compounds have been identified by FTIR and proton NMR. The prepared ester was diagnosed by the infrared spectrum as it gave a band at the frequency (1734 cm) belonging to the carbonyl ester group, the high value of the elastic carbonyl group compared to its value in carboxylic acid, but in the NMR of the proton, the disappearance for proton signal of the OH group is a clear indication of the composition of the ester. The hydrazide preparation reaction is one of the nucleophilic compensation reactions on the carbon atom in the carbonyl group, as the reaction takes place through a tetrahedral mechanism and in two stage, the 1st step is a nucleophilic attack of hydrazine on the carbonyl group, as the hybridization of the carbonyl carbon atom turns into sp3 and the formation of an unstable intermediate compound. In the 2nd stage, the intermediate compound suffered from the deletion of the ethoxy group to give hydrazide. Compound 2a was diagnosed using the infrared spectrum, where the spectrum showed a band at frequency 168a belonging to the carbonyl group and a band at frequency 3380 belonging to the stretch of the N-H group. It is noted that the frequency of stretching the carbonyl group of hydrazide is lower than at ester, and the reason for this is due to the phenomenon of resonance in hydrazide, which leads to a reduction in the characteristic for double bond (C=O) and then decreases the strength constant of this bond and decreases its frequency. Proton NMR of hydrazide shows a new signal in (8.4) ppm referring to the N-H. Compounds (3a-8a) Diagnosed using an infrared spectrum, a distinctive band appeared at the frequency (1622-1647) It belongs to the stretch of the right group and its band at (1699-1647) due to the stretch of the carbonyl group as well as its band at the frequency 3335-3445 (cm-1) It due to the stretch of the N-H group in addition to the beams resulting from the aromatic rings and the substituted on them
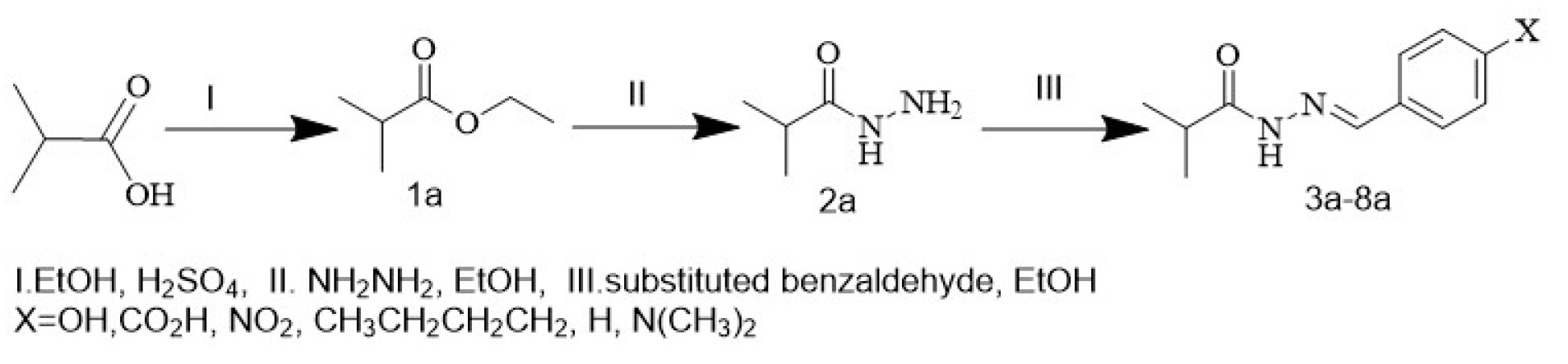
Scheme 1 explains the preparation of the compounds (1a-8a)
Molecules Library Preparation
The newly compounds (3a, 4a, 5a, 6a, 7a, and 8a) were synthesized and characterization to using in molecular docking process, Chemdraw Ultra 12.0 (https://chemdraw pro.software.informer.com/12.0/) the compounds 3D structures were created using software (3a, 4a, 5a, 6a, 7a, and 8a) and followed by energy minimization. Using Hyperchem 8.08 program, the structure was pre-optimized using the semi-empirical AM1 approach. To find the most stable conformation, the structures was optimized using the (DFT) approach utilizing the B3LYP/6-31G basis set. Maximum force, root-mean-square force, maximum displacement, and RMS displacement all converge to “YES,” which is the default value. After calculating the vibrational frequencies of the medications, all values are positive, which suggests that the drugs are stable. TO investigate ligand affinity, the optimized structures were merged into a single database on MOE program (Molecular Operating Environment, 2019) (Figure 2). Additionally, the schematic depiction of the docking process, drug analysis, and reactivity is shown in Figure 1.
Figure 1: Diagrammatic representation of drug analysis, reactivity, and the docking process
Figure 2: 2D (1a-8a) compounds
Receptors Preparation
The Protein Data Bank was used to select the protein crystal structures (8C7Y) (Figure 3). Since the water molecule in the target protein's active site is crucial, it was added to the active site to guarantee that the ligand and the target would form a hydrogen bond. The missing bonds that were broken in XRD were subsequently fixed to create the protein’s structure, and hydrogen atoms were inserted as well. Crucially, PDB is a reliable global database for biological macromolecule crystal structures.
Note
The 8C7Y protein represents the crystal structure of human tyrosinase-related protein 1 (TYRP1), a key enzyme involved in the biosynthesis of melanin. TYRP1 plays a significant role in regulating pigmentation by catalyzing the oxidation of intermediates in the melanin production pathway. Dysregulation of TYRP1 expression or activity has been implicated in several pigmentary disorders and is also associated with melanoma, a serious form of skin cancer. The 8C7Y structure includes a bound inhibitor molecule that stabilizes the active site and offers insights into ligand binding mechanisms. This protein structure serves as a valuable model for in silico screening, especially for identifying novel inhibitors targeting TYRP1 for potential therapeutic intervention in melanoma. In the present study, the ligand co-crystallized in the 8C7Y structure was considered a standard reference molecule to assess the binding affinity and interaction profile the designed test compounds.
Ligand–Protein Molecular Docking
The molecular operating environment (MOE) software was used to all docking & scoring computations (MOE,2019). The protein (8C7Y) (Figure 3) was acquired from the data bank of Protein at a resolution of (1.65) A0, For docking investigations, the resolution of 1.5 to 2.5 Å is regarded as excellent quality. It is well known that the energy score of less than or equal to -7kcal/mol and the RMSD value close to A0 are ideal. These two numbers are frequently used as standards to confirm for molecular docking.
Results
One essential technique in the drug development process is molecular docking. MOE program was used in this investigation to estimate the binding modes of the produced compounds and carry out all molecular docking compoutations (3a, 4a, 5a, 6a, 7a, and 8a) with the protein (8C7Y) (Figure 3). The predicted binding affinities and features of the investigated compounds (3a, 4a, 5a, 6a, 7a, and 8a) towards (8C7Y) are listed in Table 1, and Table 2 shows the best binding poses of compounds (3a, 4a, 5a, 6a, 7a, and 8a) against target protein. The 2D and 3D representations of interactions of the inspected compounds with the key amino acid residues of (8C7Y) protein are illustrated in the next figures and tables. Compounds (3a, 4a, 5a, 6a, 7a, and 8a) showed good binding affinity values (Table 2, Table 3) with protein (PDB ID: 8C7Y). The binding and mode of interactions of the compound (3a, 4a, 5a, 6a, 7a, and 8a) with the (PDB ID: 8C7Y) protein are shown in 2D and 3D figures. From the interactions, it has been shown that primarily there are different types of interactions (hydrogen bonding and hydrophobic interactions) the following figs. Show the interactions that was further investigated to bond lengths and hydrogen bonding in the active sites. The results of figures show the compounds (3a, 4a, 5a, 6a, 7a, and 8a) interact with different amino acid residues in different interactions: H-donor, H-acceptor, and H-pi, two H-acceptor and pi-H interactions with water and different amino acids. The energy binding and distance interaction are shown in Table 3.
Figure 3: Crystal structure of the selected protein 8C7Y
Table 1: The binding affinity and QPLD result of (1a, 2a, 3a, 4a, 5a, 6a) with the (3C7Y) protein
Compounds | pose | Binding Affinity (Kcal/mol) | RMSD (Å) | E_conf | E_place | E_score1 | E_refine | E_score2 |
1a - pose1 | 1 | -7.17741 | 1.945855 | 29.64643 | -10.6179 | -11.9379 | -30.1268 | -7.17741 |
1a - pose2 | 1 | -6.63026 | 1.257265 | 28.85591 | -10.3332 | -11.6297 | -30.5593 | -6.63026 |
1a - pose3 | 1 | -6.57624 | 1.210807 | 28.74787 | -10.2663 | -11.8296 | -29.8933 | -6.57624 |
1a - pose4 | 1 | -6.55121 | 1.426668 | 31.32655 | -10.2776 | -11.6212 | -31.4313 | -6.55121 |
1a - pose5 | 1 | -6.43826 | 0.974687 | 28.8217 | -11.5119 | -13.9712 | -28.5273 | -6.43826 |
2a - pose1 | 2 | -7.12033 | 0.843547 | 31.77107 | -11.8847 | -13.509 | -33.7478 | -7.12033 |
2a - pose2 | 2 | -7.08737 | 1.075792 | 30.20456 | -10.9446 | -12.7571 | -31.9427 | -7.08737 |
2a - pose3 | 2 | -7.55002 | 2.113482 | 32.65931 | -9.9755 | -12.7762 | -34.3514 | -7.55002 |
2a - pose4 | 2 | -7.04244 | 0.921629 | 31.64273 | -11.4647 | -12.2423 | -32.2145 | -7.04244 |
2a - pose5 | 2 | -7.03337 | 1.205919 | 32.45829 | -11.1316 | -12.4542 | -33.3514 | -7.03337 |
3a - pose1 | 3 | -7.72906 | 1.934612 | 77.52537 | -12.3667 | -10.7326 | -34.9575 | -7.72906 |
3a - pose2 | 3 | -7.1349 | 0.666221 | 76.00598 | -13.1023 | -10.1886 | -35.5252 | -7.1349 |
3a - pose3 | 3 | -7.06245 | 1.086628 | 78.55341 | -11.0983 | -9.97505 | -35.2173 | -7.06245 |
3a - pose4 | 3 | -7.02138 | 0.823818 | 78.74446 | -13.7044 | -10.1889 | -34.2991 | -7.02138 |
3a - pose5 | 3 | -6.87788 | 1.00249 | 82.67107 | -10.634 | -9.73017 | -33.8852 | -6.87788 |
4a - pose1 | 4 | -8.00211 | 1.991665 | 56.1829 | -13.5254 | -9.35096 | -38.5884 | -8.00211 |
4a - pose2 | 4 | -7.30856 | 1.231435 | 51.00625 | -10.5128 | -9.28932 | -34.8184 | -7.30856 |
4a - pose3 | 4 | -7.23028 | 2.391136 | 51.82484 | -10.0145 | -9.19023 | -34.4831 | -7.23028 |
4a - pose4 | 4 | -7.22695 | 1.343638 | 58.53328 | -12.4945 | -9.94878 | -35.483 | -7.22695 |
4a - pose5 | 4 | -7.21639 | 2.119032 | 56.46928 | -10.3689 | -9.24811 | -34.7385 | -7.21639 |
5a - pose1 | 5 | -6.58866 | 1.432 | 46.34753 | -9.54349 | -9.13708 | -29.9237 | -6.58866 |
5a - pose2 | 5 | -6.44227 | 1.653383 | 47.45815 | -10.3198 | -9.52763 | -28.8472 | -6.44227 |
5a - pose3 | 5 | -6.36453 | 0.782683 | 49.47019 | -9.75303 | -9.37375 | -30.3698 | -6.36453 |
5a - pose4 | 5 | -6.31308 | 1.035446 | 51.22636 | -11.6528 | -9.2669 | -29.7455 | -6.31308 |
5a - pose5 | 5 | -6.30312 | 0.833546 | 47.09082 | -12.1679 | -9.05014 | -25.7788 | -6.30312 |
6a - pose1 | 6 | -6.94714 | 1.0109 | 53.62997 | -12.2807 | -9.84393 | -24.6766 | -6.94714 |
6a - pose2 | 6 | -6.94492 | 1.703457 | 45.12642 | -12.4575 | -9.9909 | -36.1282 | -6.94492 |
6a - pose3 | 6 | -7.24933 | 1.975163 | 41.08992 | -14.5218 | -9.06616 | -35.6957 | -7.24933 |
6a - pose4 | 6 | -6.82618 | 1.204674 | 46.40995 | -13.0464 | -10.0606 | -26.8923 | -6.82618 |
6a - pose5 | 6 | -6.80118 | 1.572977 | 49.22615 | -11.3076 | -9.33559 | -26.8676 | -6.80118 |
Standard | std | -8.62212 | 1.924656 | 72.87959 | -39.381 | -12.2215 | -56.3795 | -8.62212 |
Table 2: Name of compounds and SMILES
Compounds | Binding Affinity (Kcal/mol) | Bond (Å) | Smile |
1a - pose1 | -7.17741 | 1.945855 | O=C(N/N=C/c1ccc(O)c1C)C(C)C |
1a - pose2 | -6.63026 | 1.257265 | O=C(N/N=C/c1ccc(O)c1C)C(C)C |
1a - pose3 | -6.57624 | 1.210807 | O=C(N/N=C/c1ccc(O)c1C)C(C)C |
1a - pose4 | -6.55121 | 1.42668 | O=C(N/N=C/c1ccc(O)c1C)C(C)C |
1a - pose5 | -6.43826 | 0.974687 | O=C(N/N=C/c1ccc(O)c1C)C(C)C |
2a - pose1 | -7.12033 | 0.843547 | O=C(N/N=C/c1ccc(C(=O)OC)c1C)C |
2a - pose2 | -7.08377 | 1.075792 | O=C(N/N=C/c1ccc(C(=O)OC)c1C)C |
2a - pose3 | -7.55002 | 2.113482 | O=C(N/N=C/c1ccc(C(=O)OC)c1C)C |
2a - pose4 | -7.04244 | 0.921269 | O=C(N/N=C/c1ccc(C(=O)OC)c1C)C |
2a - pose5 | -7.03337 | 1.205919 | O=C(N/N=C/c1ccc(C(=O)OC)c1C)C |
3a - pose1 | -7.72906 | 1.934612 | O=N+c1ccc(/N=N/C(=O)C(C)C)cc1 |
3a - pose2 | -7.1349 | 0.666221 | O=N+c1ccc(/N=N/C(=O)C(C)C)cc1 |
3a - pose3 | -7.06245 | 1.082668 | O=N+c1ccc(/N=N/C(=O)C(C)C)cc1 |
3a - pose4 | -7.02132 | 0.828136 | O=N+c1ccc(/N=N/C(=O)C(C)C)cc1 |
3a - pose5 | -6.87788 | 1.00249 | O=N+c1ccc(/N=N/C(=O)C(C)C)cc1 |
4a - pose1 | -8.00211 | 1.909165 | O=C(N/N=C/c1cccc(CCCC)c1C)C(C)C |
4a - pose2 | -7.30886 | 1.231435 | O=C(N/N=C/c1cccc(CCCC)c1C)C(C)C |
4a - pose3 | -7.23028 | 2.391134 | O=C(N/N=C/c1cccc(CCCC)c1C)C(C)C |
4a - pose4 | -7.22695 | 1.343638 | O=C(N/N=C/c1cccc(CCCC)c1C)C(C)C |
4a - pose5 | -7.21639 | 2.119032 | O=C(N/N=C/c1cccc(CCCC)c1C)C(C)C |
4a - pose1 | -6.58866 | 1.432 | O=C(N/N=C/c1ccccc1)C(C)C |
5a - pose2 | -6.44227 | 1.635383 | O=C(N/N=C/c1ccccc1)C(C)C |
5a - pose3 | -6.36453 | 0.782683 | O=C(N/N=C/c1ccccc1)C(C)C |
5a - pose4 | -6.31308 | 1.035446 | O=C(N/N=C/c1ccccc1)C(C)C |
5a - pose5 | -6.30312 | 0.833546 | O=C(N/N=C/c1ccccc1)C(C)C |
6a - pose1 | -6.94714 | 1.0109 | O=C(N/N=C/c1cccn(C)c1)C(C)C |
6a - pose2 | -6.94492 | 1.703457 | O=C(N/N=C/c1cccn(C)c1)C(C)C |
6a - pose3 | -7.24933 | 1.975163 | O=C(N/N=C/c1cccn(NC)c1C)C |
6a - pose4 | -6.82618 | 1.204674 | O=C(N/N=C/c1cccn(C)c1)C(C)C |
6a - pose5 | -6.80118 | 1.572977 | O=C(N/N=C/c1cccn(C)c1)C(C)C |
Standard | -8.62212 | 1.924656 | Clc1cnc2[nH]c(C(=O)c3c(F)c(Nc(=O)c4cc(C(N)=O)cc(C)c4)cc3F)cc2c1 |
Table 3: Details of the best pose of protein 8C7Y
| Compounds | Binding Affinity (Kcal/mol) | RMSD (Å) | Atom of compound | Atom of Receptor | Involved receptor residues | Type of interaction bond | Distance (Å) | E (kcal/mol) |
1a - pose1 | -7.17741 | 1.945855 | N 5; O 4 | OG1; O | THR 529; HOH 913 | (B) H-donor; (B) H-acceptor | 3.41; 2.87 | -0.6; -0.9 |
2a - pose3 | -7.55002 | 2.113482 | O 17; O 17; O 4 | O; O; O | CYS 532; CYS 532; HOH 913 | (B) H-donor; (B) H-donor; (B) H-acceptor | 2.97; 2.97; 2.72 | -2.6; -2.6; -0.8 |
3a - pose1 | -7.72906 | 1.934612 | O 4; O 4 | CA; CA | PHE 595; PHE 595 | (B) H-acceptor; (B) H-acceptor | 3.54; 3.54 | -0.7; -0.7 |
4a - pose1 | -8.00211 | 1.991665 | N 5; C 7; C 15; C 15; 6-ring | OE2; OE2; 5-ring; 5-ring; CD2 | GLU 501; GLU 501; HIS 574; HIS 574; LEU 505 | (B) H-donor; (B) H-donor; (B) H-pi; (B) H-pi; (B) pi-H | 3.32; 3.08; 4.10; 4.09; 4.39 | -2.2; -0.8; -0.8; -0.7; -0.6 |
5a - pose2 | -6.44227 | 1.635383 | 5-ring | 6-ring | P54 1 | (A) pi-pi | 1.01 | -1.0 |
6a - pose3 | -7.24933 | 1.975163 | N 5; N 5 | OG1; O | THR 529; THR 529 | (B) H-donor; (B) H-donor | 3.09; 3.00 | -1.8; -2.1 |
Standard | -8.62212 | 1.924656 | N1 1; N2 6; O1 10; O3 16 | OE2; GLN 530; HOH 913; CYS 532 | GLU 501; GLN 530; HOH 913; CYS 532 | (B) H-donor; (B) H-donor; (B) H-acceptor; (B) H-acceptor | 2.84; 2.87; 2.68; 2.91 | -1.5; -5.0; -2.4; -5.3 |
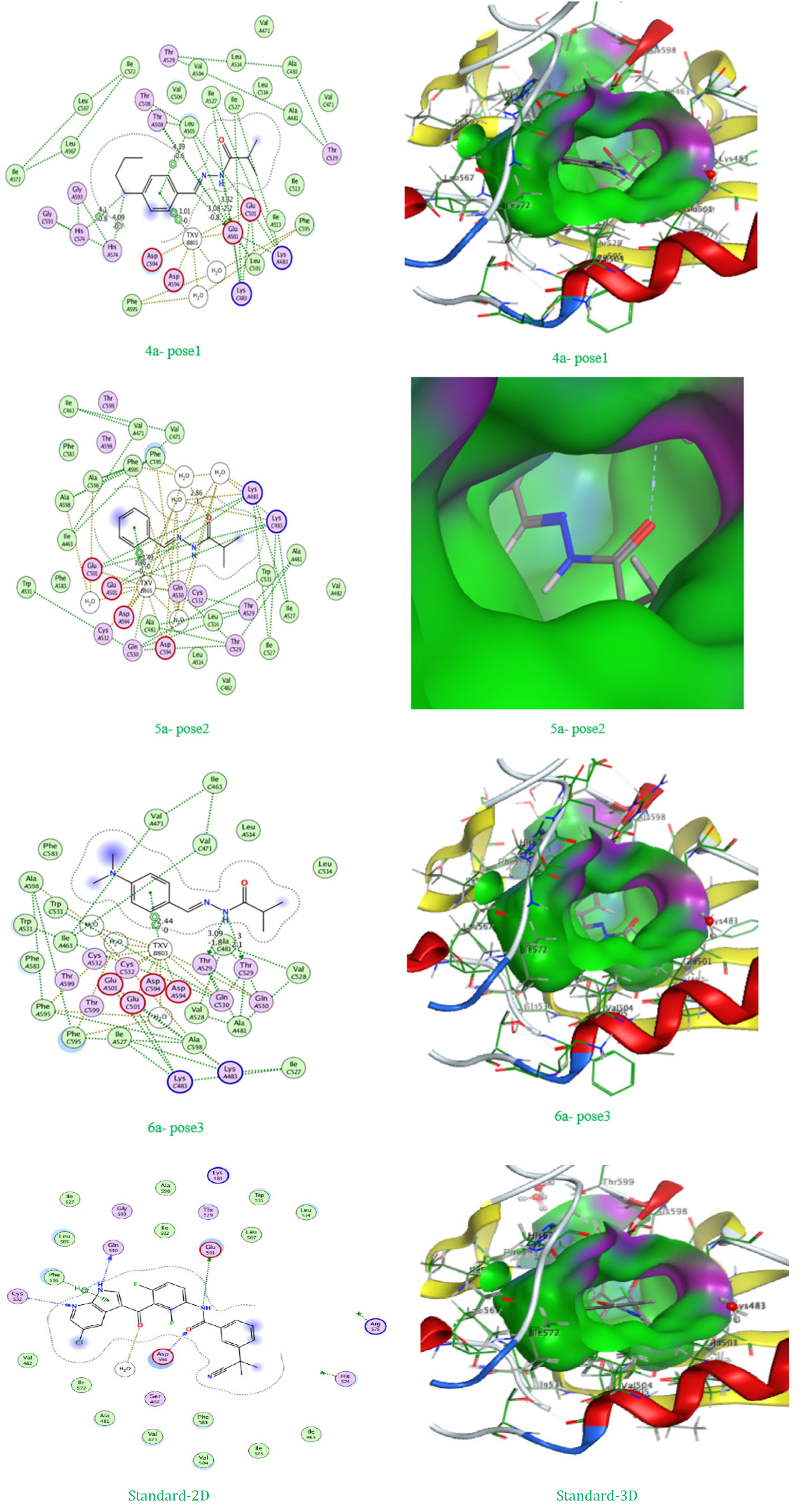
Figure 4: 2D and 3D of the best pose
The docking study for compounds 3a, 4a, 5a, 6a, 7a, and 8a against the 8C7Y protein, compared to the standard TXV, reveals important insights not only about their binding affinities and RMSD values but also regarding their modes of interaction and the role of specific amino acid residues in complex stabilization. Starting with the binding affinities, the standard compound TXV again exhibited the highest affinity at -8.62212 kcal/mol, setting a strong benchmark. Among the new ligands, compound 53 (pose 1) came closest with a binding affinity of -8.00211 kcal/mol, followed by compound 5a (pose 1) at -7.72906 kcal/mol, and compounds 4a, 5a, and 3a with best poses ranging between approximately -7.17 to -7.55 kcal/mol. The rest of the compounds had moderately less negative values, reflecting lower binding strength. Examining the RMSD values for the best poses, all are generally within the acceptable range for docking studies, with most top poses around 1.6–2.1 Å. This demonstrates that the predicted binding conformations are stable and reliable, making the energy results more trustworthy.
Delving into the interaction details and amino acid involvement, the results highlight key differences among the compounds. For example, compound 6a (pose 1) forms strong hydrogen bonds with GLU 501 (distances 3.32 Å and 3.08 Å), and unique pi-H interactions with HIS 574 (distances 4.10 Å and 4.09 Å) as well as a pi-H interaction with LEU 505 (4.39 Å). These multiple interactions not only explain the high binding affinity but also show how the ligand is effectively anchored in the active site through both polar (hydrogen bonding) and hydrophobic (aromatic) contacts. The energy contribution of these interactions varies, with hydrogen bonds ranging from -2.2 to -0.8 kcal/mol and pi-H bonds at -0.8 to -0.6 kcal/mol. For compound 3a (pose 1), hydrogen bonds are formed with THR 529 (3.41 Å, -0.6 kcal/mol) and a water molecule HOH 913 (2.87 Å, -0.9 kcal/mol), which, while moderate in energy, still provide a stabilizing effect. Compounds 4a (pose 3) and 8a (pose 3) both engage in hydrogen bonding with CYS 532 and THR 529 respectively, with bond energies between -2.6 and -1.8 kcal/mol and bond distances from 2.97 to 3.09 Å, underscoring the role of cysteine and threonine residues in ligand stabilization.
Compound 5a (pose 1) stands out for its interactions with PHE 595, where it forms two nearly identical H-acceptor contacts (both 3.54 Å, -0.7 kcal/mol). Compound 7a (pose 2) interacts with residue P54 1 via a pi-pi stacking interaction at a distance of 1.01 Å and an energy of -1.0 kcal/mol, which highlights the importance of π-stacking in some ligand-protein recognition scenarios.
The standard compound TXV exhibits a highly cooperative binding mode, with hydrogen bonds to GLU 501 (2.84 Å, -5.5 kcal/mol), GLN 530 (2.87 Å, -5.0 kcal/mol), and water HOH 913 (2.68 Å, -2.4 kcal/mol), and additional H-acceptor and H-bonding with CYS 532 (2.91 Å, -5.3 kcal/mol). These short and energetically strong bonds (up to -5.5 kcal/mol) explain the superior affinity of TXV, as it achieves deep embedding into the active site via multiple points of contact—primarily through strong polar interactions.
A comprehensive comparison shows that, while the new compounds do not entirely match the binding energy or interaction richness of TXV, some approach it closely, particularly in their use of multiple hydrogen bonds and aromatic contacts with essential residues like GLU 3a1, HIS 574, PHE 595, THR 5a9, and CYS 6a2. The distances for most hydrogen bonds remain within the optimal range (2.7–3.5 Å), ensuring their structural and energetic significance.
In summary, these results suggest that compound 6a (pose 1), due to its combination of strong hydrogen bonds and favorable aromatic interactions with critical residues, is the most promising among the tested ligands. Other compounds like 5a, 4a, and 8a also display significant binding potential due to their diverse interaction profiles. However, the standard TXV still outperforms in both binding strength and interaction quality, emphasizing the need for further ligand optimization if the goal is to exceed its inhibitory efficiency. The detailed consideration of interaction type, energy, and involved amino acids provides a deeper mechanistic understanding of binding, which is essential for rational drug design and further experimental follow-up.
Figure 5: Best binding affinity for each compound against 8C7Y
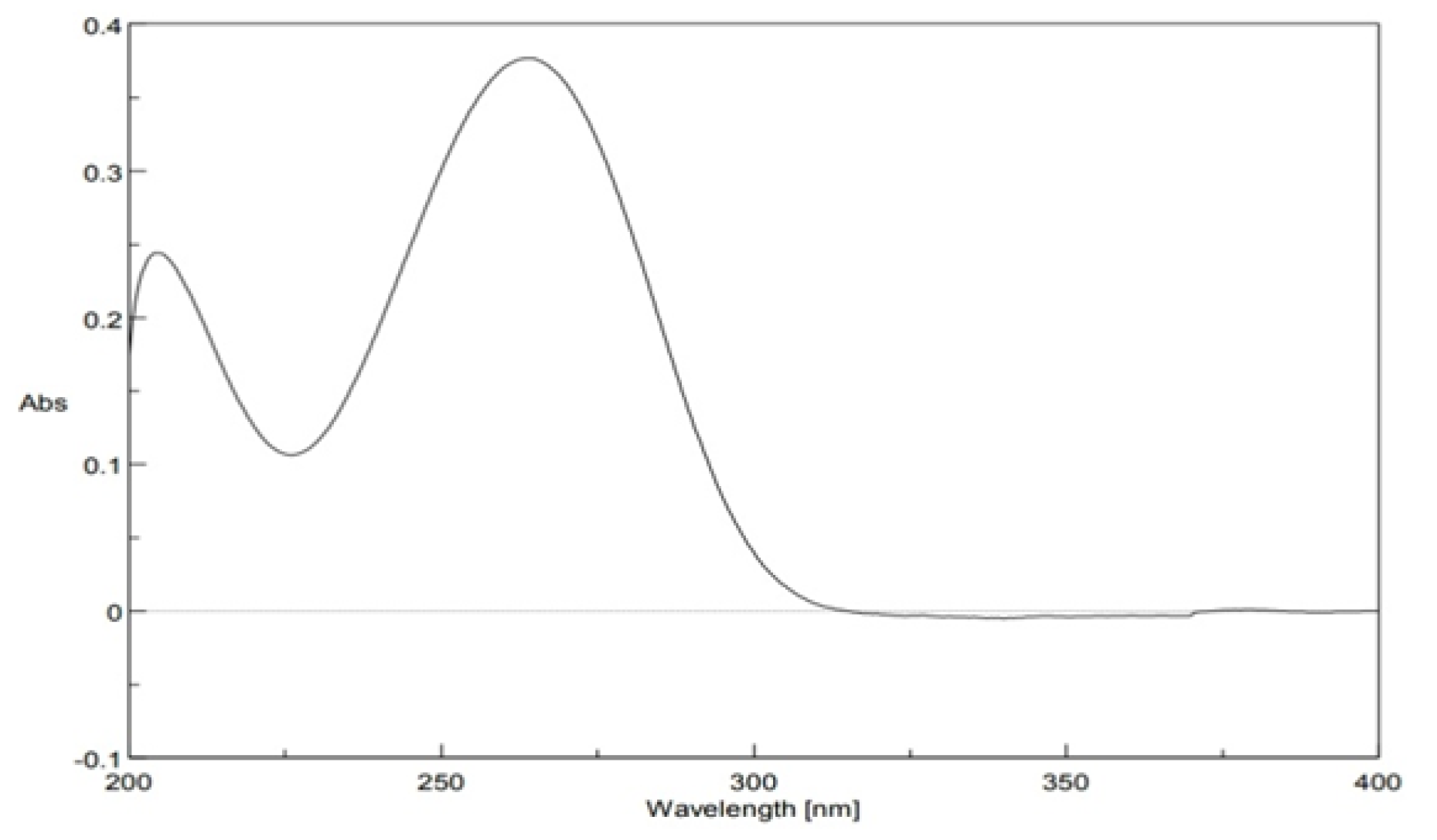
Figure 6: RMSD of best pose for each compound against 8C7Y
Figure 7: Docking results: compound 50-55 vs TXV (8C7Y)
Figure 5 shoes the best binding affinity values (in kcal/mol) of six test compounds (3a to 8a) in comparison with the reference compound TXV against the 8C7Y protein. Compound 6a demonstrated the most favorable binding energy among the test compounds, approaching the affinity of the TXV standard. Lower binding affinity values indicate stronger interactions between the ligand and the protein, with TXV showing the highest binding strength (–8.62 kcal/mol), followed closely by compound 6a (–8.00 kcal/mol).
Figure 6 compares the RMSD (Root Mean Square Deviation) values for the best docking poses of each compound. RMSD provides insight into the stability and accuracy of the docking pose. All compounds, including TXV, show RMSD values below 2.2 Å, indicating good structural alignment. Compound 7a shows the most compact binding pose (lowest RMSD), while compound 4a has a slightly higher deviation, suggesting more conformational variation during docking.
Figure 7 shows the binding affinity (in kcal/mol, solid lines) and RMSD values (in Å, dashed lines) across five docking poses for each of the compounds 3a–8a, compared with the standard compound TXV, all docked against the 8C7Y protein. Lower binding affinity values (more negative) represent stronger interactions, and lower RMSD values indicate more stable and consistent docking poses. The compound 6a shows the best affinity (−8.00 kcal/mol), closely approaching the standard TXV (−8.62 kcal/mol). The RMSD trends highlight the conformational flexibility of each compound, where compound 7a has relatively high RMSD variance, while TXV remains stable throughout. This dual representation helps evaluate both binding strength and structural stability simultaneously.
Hydrazone compounds are successfully manufactured under conventional working conditions with a productivity ranging from medium to high. Spectral data have confirmed the expected compositions of the compounds and have been thoroughly tested by molecular docking, and have given good results.
Jassim, H.Y., and M.K. Hadi. “Synthesis, Characterization and Preliminary Anti-Microbial Evaluation of New Flurbiprofen Hydrazide Derivatives.” Iraqi Journal of Pharmaceutical Sciences, University of Baghdad - College of Pharmacy, 2024, pp. 31–42. doi: 10.31351/vol33iss(4SI)pp31-42.
Ismail, M., et al. “Synthesis of Hydrazone-Based Polyhydroquinoline Derivatives - Antibacterial Activities, α-Glucosidase Inhibitory Capability, and DFT Study.” RSC Advances, vol. 14, no. 16, April 2024, pp. 10978–10994. doi: 10.1039/d4ra00045e.
Çakmak, R., et al. “Synthesis of Novel Hydrazide-Hydrazone Compounds and In Vitro and In Silico Investigation of Their Biological Activities Against AChE, BChE, and hCA I and II.” ACS Omega, vol. 9, no. 18, May 2024, pp. 20030–20041. doi: 10.1021/acsomega.3c10182.
Taha, L.L., and M.M.J. Al-Mudhafar. “Synthesis, Characterization, Molecular Docking, ADME Study, and Antimicrobial Evaluation of New Fenamate-Isatin Hybrids.” Pharmacia, vol. 72, 2025. doi: 10.3897/pharmacia.72.e143889.
Koç, H.C., et al. “Synthesis of Some Novel Hydrazide-Hydrazones Derived from Etodolac as Potential Anti-Prostate Cancer Agents.” Journal of Research in Pharmacy, vol. 26, no. 1, 2022, pp. 1–12. doi: 10.29228/jrp.97.
Ali, Yousef Sabah, et al. “Design, Molecular Docking, Synthesis, Characterization and Preliminary Evaluation of Novel 1,3,4-Oxadiazole Derivatives as Cyclin-Dependent Kinase 2 Inhibitors.” Al Mustansiriyah Journal of Pharmaceutical Sciences, vol. 25, no. 1, January 2025, pp. 94–109. doi: 10.32947/ajps.v25i1.1132.
Jasril, et al. “Synthesis, In Vitro Antioxidant Activity, and Toxicity Evaluation of Hydrazone Derivatives Naphthalene-1-ylmethylene Hydrazine.” Journal of Physics: Conference Series, IOP Publishing Ltd, October 2021. doi: 10.1088/1742-6596/2049/1/012050.
Sonu, et al. “Synthesis, Characterization, Molecular Docking and Pharmacological Evaluation of Isoxazole Derivatives as Potent Anti-Inflammatory Agents.” Heliyon, vol. 10, no. 22, November 2024. doi: 10.1016/j.heliyon.2024.e40300.
Daoud, K.M., et al. “Synthesis of 2-(3-Chloro-4-Nitro-1-Benzothien-2-yl)-1,3,4-Oxadiazole-1,3,4-Thiadiazole and 5-(3-Chloro-4-Nitro-1-Benzothien-2-yl)-4H-1,2,4-Triazole-3-Thiol.”
Boudissa, R., et al. “Synthesis, Characterization, DFT, Antibacterial, ADME-T Properties, and Molecular Docking of New N-Functionalized Thiazolidinones.” Journal of Molecular Structure, 2024, p. 138004. doi: 10.1016/j.molstruc.2024.138004.
Yadav, C.S., et al. “Synthesis, Characterization, Quantum Chemical Modelling, Molecular Docking, In Silico and In Vitro Assessment of 3-(2-Bromo-5-Fluorophenyl))-1-(Thiophen-2-yl)prop-2-en-1-one.” Scientific Reports, vol. 14, no. 1, December 2024. doi: 10.1038/s41598-024-79747-8.
Sonu, et al. “Synthesis, Characterization, Molecular Docking and Pharmacological Evaluation of Isoxazole Derivatives as Potent Anti-Inflammatory Agents.” Heliyon, vol. 10, no. 22, November 2024. doi: 10.1016/j.heliyon.2024.e40300.
Ahmed, A.A., et al. “Synthesis and Characterization of Few New Substituted 1,3,4-Oxadiazoles, 1,2,4-Triazoles and Schiff Bases via Chalcone Compounds.” International Journal of Drug Delivery Technology, vol. 12, no. 3, July 2022, pp. 1087–1092. doi: 10.25258/ijddt.12.3.27.
Daoud, K.M., and H.S. Aziz. “Synthesis of 2-Isobutyl, 2-Isobutyl-5-Aryl and 2-Isobutyl-5-Thiol-1,3,4-Oxadiazoles.”
Alotabi, S.H. “Synthesis, Characterization, Anticancer Activity, and Molecular Docking of Some New Sugar Hydrazone and Arylidene Derivatives.” Arabian Journal of Chemistry, vol. 13, no. 3, March 2020, pp. 4771–4784. doi: 10.1016/j.arabjc.2019.12.006.
Daoud, K.M., et al. “Synthesis of 2-Benzamidomethyl-5-Substituted Amino-1,3,4-Thiadiazoles, 2,5-Disubstituted 1,3,4-Oxadiazole and 4,5-Disubstituted 1,2,4-Triazole-3-Thiol.”
El-Din, N.S., and A. Barseem. “Synthesis, Bioactivity and Docking Study of Some New Indole-Hydrazone Derivatives.” Journal of Applied Pharmaceutical Science, vol. 6, no. 12, 2016, pp. 075–083. doi: 10.7324/JAPS.2016.601211.
Taha, L.L., and M.M.J. Al-Mudhafar. “Synthesis, Characterization, Molecular Docking, ADME Study, and Antimicrobial Evaluation of New Fenamate-Isatin Hybrids.” Pharmacia, vol. 72, 2025. doi: 10.3897/pharmacia.72.e143889.
